CUTANA™ Fiber-seq Kit
Gain multiomic insights with a single long-read-sequencing assay
Product Description
Fiber-seq simultaneously profiles chromatin accessibility, DNA methylation, and genetic variants, consolidating what typically requires three or more separate assays into one long-read sequencing (LRS) assay.
Fiber-seq is a breakthrough approach that simultaneously maps genetic and epigenetic features from individual chromatin fibers. This single assay delivers insights that would normally require integrating data from multiple methods such as ATAC-seq, ChIP-seq, and bisulfite sequencing. This kit includes reagents to go from cells to purified genomic DNA that can be used directly in either Pacific Biosciences® (PacBio® HiFi Sequencing) or Oxford Nanopore Technologies® (ONT® Nanopore Sequencing) library preparation workflows. The recommended input for Fiber-seq is 1,000,000 native human nuclei per reaction. The Fiber-seq Kit has been successfully applied to cryopreserved and lightly cross-linked nuclei.
Benefits
Comprehensive solution – This kit includes key reagents needed to go from cells to library prep-ready DNA.
User-friendly resources – The User Manual is complete with a detailed protocol, QC checks, FAQs, and expert tips to get you started, while the bench-proof Quick-Start Card provides a concise protocol for convenient reference.
Easy workflow integration – Fiber-seq can be performed in <2 hours directly upstream of routine long-read sequencing library preparation and uses reagents vetted for compatibility with library prep kits from major LRS platforms.
Powered by Hia5 6mA methyltransferase – Hia5 is the highest activity 6mA-methyltransferase available and is exclusive to CUTANA Fiber-seq Kits. This more active enzyme enables faster incubations, which results in better resolution of protein footprints [2-4].
Stringent quality control – Each new lot undergoes rigorous quality control checks to ensure components yield consistent results in your research.
Questions?
Connect with us today to get started on your Fiber-seq sequencing experiment.
Fiber-seq workflow
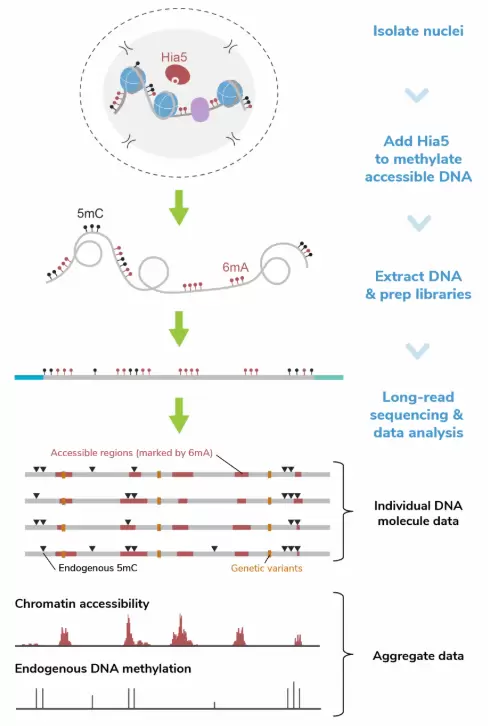
Figure 1: In the Fiber-seq workflow, isolated nuclei are incubated with Hia5 N⁶-methyladenine methyltransferase (6mA MTase) and cofactor S-Adenosylmethionine (SAM) to selectively methylate adenines within regions of accessible chromatin. After a brief 10-minute labeling step, the reaction is quenched and genomic DNA (gDNA) is purified and prepared for long-read sequencing following standard native DNA sequencing protocols. Long-read sequencing data is next analyzed at a per molecule level to observe heterogeneity in chromatin structure within a sample or in aggregate to identify sample-wide trends in accessibility and DNA methylation.
Applications
- Chromatin accessibility
- DNA methylation (5mC, 5hmC)
- Protein footprints (including nucleosomes, transcription factors, and RNA polymerase)
- Genetic variants (SNPs, CNVs, and structural variants)
Performance Data

Figure 2: Fiber-seq simultaneously captures chromatin accessibility and endogenous DNA methylation
CUTANA™ Fiber-seq simultaneously detects open chromatin (marked by N⁶-methyladenine; 6mA) and DNA methylation (5mC) in human leukemia (K562) cells. The top Fiber-seq track displays aggregate 6mA signal. ATAC-seq and CTCF CUT&RUN data are shown for comparison. CTCF is a DNA-binding protein that regulates chromatin structure and is often enriched at open chromatin regions. Each horizontal line in the Fiber-seq panel represents a single long read, providing single-DNA molecule data from 60 individual chromatin fibers.

Figure 3: Fiber-seq reveals protein footprints genome wide and at higher resolution than ATAC-seq.
(A) Fiber-seq data from 30 individual DNA molecules are shown, with the top track displaying a pileup of 6mA signal marking open chromatin. Each horizontal line below this track represents a single DNA molecule. Dotted boxes indicate CTCF motif regions. Because Fiber-seq provides single-molecule resolution, heterogeneity in protein binding across the cell population is revealed (the leftmost boxed region shows variable 6mA labeling, suggesting differential CTCF occupancy).
(B) Genome-wide 6mA signal from this Fiber-seq experiment demonstrates robust footprinting of CTCF sites. Elevated 6mA levels flanking the motif midpoint reflect accessible DNA surrounding the CTCF binding sites, which is protected from 6mA labeling.

Figure 4: Fiber-seq captures chromatin accessibility, DNA methylation, and inferred nucleosome positioning genome-wide from a single experiment.
Heatmaps show aligned single-molecule data for 6mA signal, nucleosome positioning, and endogenous 5mC levels centered at transcription start sites (TSSs). TSSs enriched for 6mA display well-defined nucleosome organization and a depletion of 5mC, consistent with active promoter regions. These patterns align with CUT&RUN profiles for histone modifications, including H3K4me3 (active TSS) and H3K27me3 (repressed genes).
Any Questions Left?
Contact us for assistance to get started on your Fiber-seq sequencing experiment.
Want to learn more about Fiber-seq?
View the Science publication Single-molecule regulatory architectures captured by chromatin fiber sequencing, A.B.Stergachis, et al. (2020).
